¶ Los Marcadores del Envejecimiento
¶ Descripción general
Los Marcadores del Envejecimiento (Hallmarks of Aging) constituyen el marco científico definitivo para comprender los mecanismos biológicos que impulsan el proceso de envejecimiento. Propuesto originalmente en un artículo histórico de 2013 por López-Otín et al., el marco definía inicialmente nueve marcadores. En 2023, se publicó una actualización importante en Cell, ampliando el universo de los marcadores del envejecimiento a doce procesos distintos e interconectados[1][2].
Estos marcadores proporcionan un lenguaje común para la investigación del envejecimiento e identifican dianas específicas para intervenciones destinadas a prolongar el periodo de salud y la esperanza de vida.
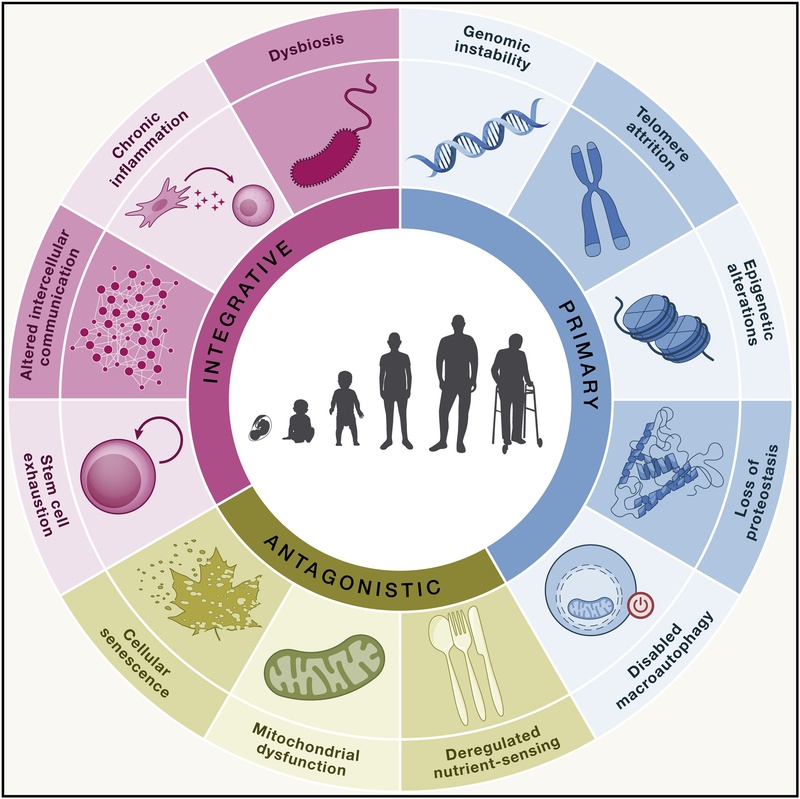
Figura 1: Los 12 Marcadores del Envejecimiento, categorizados en grupos Primarios, Antagónicos e Integradores. Adaptado de López-Otín et al. (2023).
¶ Evolución del marco de trabajo
La clasificación original de 2013 estableció la investigación del envejecimiento como una disciplina molecular rigurosa. La actualización de 2023 incorpora una década de nuevos descubrimientos, añadiendo tres nuevos marcadores: Macroautofagia deteriorada (Disabled Macroautophagy), Inflamación crónica (Chronic Inflammation) y Disbiosis (Dysbiosis)[1:1].
¶ Criterios para un marcador
Para que un proceso biológico sea calificado como un marcador del envejecimiento, debe cumplir estrictamente tres condiciones[1:2]:
- Manifestación asociada a la edad: El fenómeno debe observarse durante el envejecimiento normal.
- Aceleración experimental: La intensificación experimental del proceso debe acelerar el envejecimiento.
- Potencial terapéutico: La inhibición o mejora terapéutica del proceso debe retrasar el envejecimiento normal.
¶ Los 12 marcadores del envejecimiento
Los marcadores se organizan jerárquicamente en tres categorías: Primarios (causas del daño), Antagónicos (respuestas al daño) e Integrativos (culpables del fenotipo).
¶ Marcadores primarios (Causas del daño)
Estos son los desencadenantes iniciales del proceso de envejecimiento, caracterizados por la acumulación de daño celular.
¶ 1. Inestabilidad genómica
El envejecimiento se acompaña de una acumulación de daño genético a lo largo de la vida. Esto incluye mutaciones somáticas, aneuploidías cromosómicas y variaciones en el número de copias. Las fuentes de daño incluyen agentes externos (UV, radiación) y amenazas internas (ROS, errores de replicación). Las deficiencias en los mecanismos de reparación del ADN (como NER, BER y reparación de DSB) aceleran el envejecimiento, como se observa en síndromes progeroides como el síndrome de Werner[1:3][2:1].
¶ 2. Desgaste de los telómeros
Los telómeros son las capas protectoras en los extremos de los cromosomas. Debido al "problema de la replicación final", los telómeros se acortan con cada división celular. Cuando alcanzan una longitud crítica, las células entran en senescencia o apoptosis. La deficiencia de telomerasa está vinculada a enfermedades de envejecimiento prematuro (disqueratosis congénita), mientras que la activación de la telomerasa puede retrasar el envejecimiento en ratones[1:4].
¶ 3. Alteraciones epigenéticas
El envejecimiento implica cambios en el panorama epigenético: modificaciones en el ADN y las histonas que regulan la expresión génica sin alterar la secuencia. Los cambios clave incluyen la hipometilación global del ADN, la hipermetilación focal en las islas CpG y los desequilibrios en la modificación de las histonas (p. ej., aumento de H4K20me3, disminución de H3K9me3). Estas alteraciones conducen al ruido transcripcional y a una expresión génica aberrante[1:5][3].
¶ 4. Pérdida de la proteostasis
La proteostasis (homeostasis proteica) asegura el correcto plegamiento y la eliminación de proteínas. El envejecimiento deteriora la red de proteostasis, lo que conduce a la acumulación de proteínas mal plegadas o agregadas (p. ej., amiloide-beta, tau). Los mecanismos clave incluyen el fallo de las chaperonas y la disfunción del sistema ubiquitina-proteasoma[1:6][4].
¶ 5. Macroautofagia desactivada (Nuevo en 2023)
La macroautofagia es el mecanismo principal para el reciclaje de orgánulos dañados y contenido citoplasmático. Aunque originalmente se incluía dentro de la proteostasis, ahora es un sello distintivo independiente. La autofagia disminuye con la edad, lo que lleva a la acumulación de "basura celular" (lipofuscina, mitocondrias dañadas). Potenciar la autofagia (p. ej., mediante rapamicina o espermidina) prolonga de manera consistente la esperanza de vida en organismos modelo[1:7][5].
¶ Sellos distintivos antagónicos (Respuestas al daño)
Estos sellos distintivos suelen comenzar como respuestas protectoras al daño primario, pero se vuelven perjudiciales cuando se vuelven crónicos o se exacerban.
¶ 6. Detección de nutrientes desregulada
La red de vías de detección de nutrientes —que incluye mTOR, AMPK, Sirtuinas e IGF-1— regula el crecimiento celular y el metabolismo. En el envejecimiento, la señalización anabólica (crecimiento) (mTOR/IGF-1) suele estar hiperactiva, mientras que la señalización catabólica (reparación) (AMPK/Sirtuinas) disminuye. La restricción calórica y los inhibidores de mTOR (rapamicina) se encuentran entre las intervenciones de longevidad más robustas conocidas[1:8][6].
¶ 7. Disfunción mitocondrial
Las mitocondrias generan energía (ATP) pero también producen especies reactivas de oxígeno (ROS). El envejecimiento conduce a una disminución de la eficiencia mitocondrial, una reducción de la biogénesis y la acumulación de mutaciones en el mtDNA. La actualización de 2023 enfatiza que las ROS no son estrictamente perjudiciales, sino que actúan como moléculas de señalización (mitohormesis); la disfunción surge cuando esta señalización se interrumpe o cuando falla el control de calidad mitocondrial (mitofagia)[1:9].
¶ 8. Senescencia Celular
La senescencia es un estado de parada estable del ciclo celular inducido por el estrés (p. ej., daño en el ADN, acortamiento de los telómeros). Si bien es esencial para la supresión de tumores y la cicatrización de heridas, la acumulación de células senescentes en los tejidos envejecidos impulsa la patología a través del Fenotipo Secretor Asociado a la Senescencia (SASP)—un cóctel de citoquinas proinflamatorias y proteasas que daña el tejido vecino[1:10][7].
¶ Sellos Integrativos (Culpables del Fenotipo)
Estos sellos surgen cuando el daño acumulado y las respuestas antagónicas superan la homeostasis tisular, lo que conduce al declive funcional.
¶ 9. Agotamiento de las Células Madre
El potencial regenerativo de los tejidos disminuye con la edad debido al agotamiento o la disfunción de las células madre adultas. Esto es impulsado por todos los sellos anteriores (p. ej., daño en el ADN en las células madre hematopoyéticas, senescencia en las células satélite). El resultado es un fallo en el mantenimiento y la reparación de los tejidos, lo que contribuye a la anemia, la sarcopenia y la inmunosenescencia[1:11].
¶ 10. Comunicación Intercelular Alterada
El envejecimiento altera las señales de comunicación entre las células (endocrinas, paracrinas y neuronales). Esto conduce a un entorno sistémico "ruidoso" caracterizado por una inflamación crónica (inflammaging) y defectos en la red neuro-endocrino-inmune. Estrategias como la parabiosis (intercambio de sangre joven) sugieren que la restauración de los factores de señalización juveniles puede rejuvenecer los tejidos envejecidos[1:12].
¶ 11. Inflamación Crónica (Nuevo en 2023)
A menudo denominada "inflammaging", se trata de una inflamación estéril, de bajo grado y persistente que se acumula con la edad. A diferencia de la inflamación aguda (una respuesta de defensa), la inflamación crónica causa daños tisulares sistémicos. Es impulsada por el SASP de las células senescentes, los restos celulares (DAMPs) y la desregulación del sistema inmunitario (inmunosenescencia). Es un factor clave de la multimorbilidad, incluyendo las enfermedades cardiovasculares y la neurodegeneración[1:13][8].
¶ 12. Disbiosis (Novedad en 2023)
El microbioma intestinal experimenta cambios significativos durante el envejecimiento, perdiendo diversidad y desplazándose hacia un perfil proinflamatorio (p. ej., aumento de Proteobacteria). Esta "disbiosis" compromete la barrera intestinal ("leaky gut"), permitiendo que los productos bacterianos entren en la circulación y desencadenen una inflamación sistémica. Restaurar un microbioma juvenil (p. ej., mediante trasplante fecal o probióticos) puede prolongar la vida útil en modelos como el killifish[1:14][9].
¶ Interconexión y Terapéutica
Los sellos distintivos (hallmarks) no son compartimentos aislados, sino una red compleja e interactiva. Por ejemplo:
- La inestabilidad genómica puede desencadenar la senescencia celular.
- La senescencia produce SASP, impulsando la inflamación crónica.
- La inflamación perjudica la función de las células madre.
- La autofagia deficiente impide la eliminación de mitocondrias dañadas (fallo de la mitofagia), lo que provoca una mayor inflamación (activación del inflamasoma).
Esta interconectividad implica que actuar sobre un sello distintivo a menudo impacta en otros. Por ejemplo, los senolíticos (que eliminan las células senescentes) no solo reducen la senescencia, sino que también disminuyen la inflamación crónica y mejoran la función de las células madre[1:15].
¶ References
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243-278. DOI: 10.1016/j.cell.2022.11.001 ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194-1217. DOI: 10.1016/j.cell.2013.05.039 ↩︎ ↩︎
Sen, P., Shah, P. P., Nativio, R., & Berger, S. L. (2016). Epigenetic mechanisms of longevity and aging. Cell, 166(4), 822-839. ↩︎
Kaushik, S., & Cuervo, A. M. (2015). Proteostasis and aging. Nature Medicine, 21(12), 1406-1415. ↩︎
Hansen, M., Rubinsztein, D. C., & Walker, D. W. (2018). Autophagy as a promoter of longevity: insights from model organisms. Nature Reviews Molecular Cell Biology, 19(9), 579-593. ↩︎
Fontana, L., & Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell, 161(1), 106-118. ↩︎
Gorgoulis, V., et al. (2019). Cellular senescence: defining a path forward. Cell, 179(4), 813-827. ↩︎
Franceschi, C., & Campisi, J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 69(Suppl_1), S4-S9. ↩︎
Ralli, C., et al. (2022). The gut microbiome and healthy aging. Nature Reviews Gastroenterology & Hepatology. ↩︎