¶ 衰老的标志
¶ 概述
衰老的标志(Hallmarks of Aging)是理解驱动衰老过程的生物学机制的权威科学框架。该框架最初由 López-Otín 等人在2013年的一篇里程碑式论文中提出,最初定义了九个标志。2023年,《细胞》(Cell)杂志发表了一项重大更新,将衰老标志的范畴扩展到了12个截然不同且相互关联的过程[1][2]。
这些标志为衰老研究提供了一种通用语言,并为旨在延长健康寿命和绝对寿命的干预措施确定了具体靶点。
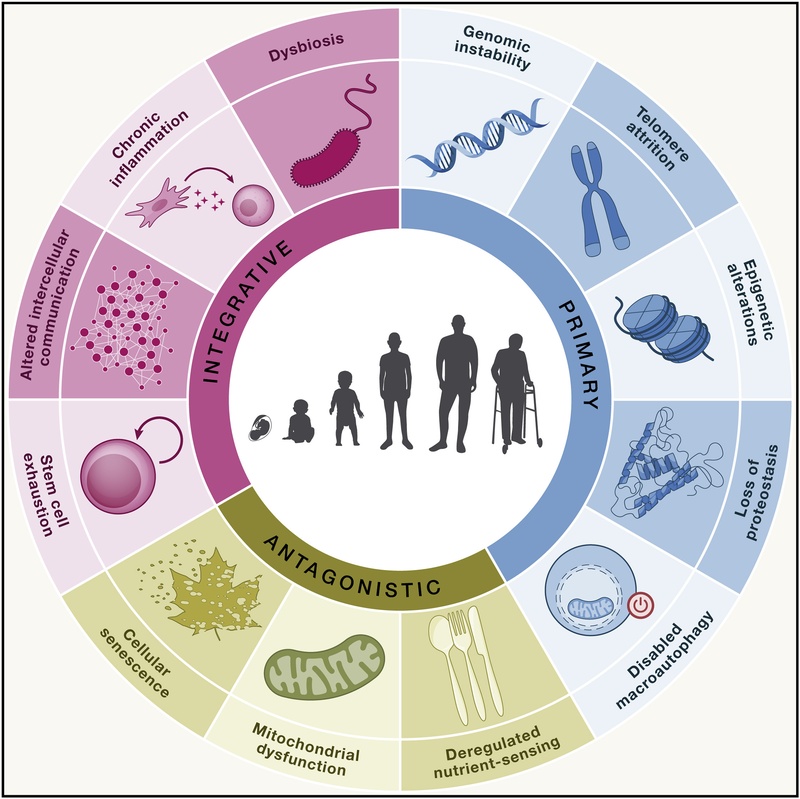
图1:衰老的12个标志,分为原发性(Primary)、拮抗性(Antagonistic)和综合性(Integrative)三类。改编自 López-Otín 等人(2023年)。
¶ 框架的演变
2013年的最初分类将衰老研究确立为一门严谨的分子学科。2023年的更新结合了十年来取得的新发现,增加了三个新标志:巨自噬失活(Disabled Macroautophagy)、慢性炎症(Chronic Inflammation)和微生态失调(Dysbiosis)[1:1]。
¶ 标志的判定标准
一个生物学过程要符合衰老标志的条件,必须严格满足以下三个条件[1:2]:
- 与年龄相关的表现(Age-associated manifestation):该现象必须在正常衰老过程中被观察到。
- 实验性加速(Experimental acceleration):在实验中强化该过程必须能够加速衰老。
- 治疗潜力(Therapeutic potential):对该过程进行治疗性抑制或改善必须能够延缓正常衰老。
¶ 衰老的 12 个标志
这些标志按层级分为三类:原发性(Primary,损伤的原因)、拮抗性(Antagonistic,对损伤的反应)和综合性(Integrative,表型的罪魁祸首)。
¶ 原发性标志(损伤的原因)
这些是衰老过程的初始触发因素,其特征是细胞损伤的积累。
¶ 1. 基因组不稳定性
衰老伴随着一生中遗传损伤的积累。这包括体细胞突变、染色体非整倍体和拷贝数变异。损伤的来源包括外部因素(紫外线、辐射)和内部威胁(ROS、复制错误)。DNA 修复机制(如 NER、BER 和 DSB 修复)的缺陷会加速衰老,正如在沃纳综合征(Werner syndrome)等早衰综合征中所见[1:3][2:1]。
¶ 2. 端粒损耗
端粒是染色体末端的保护帽。由于“末端复制问题”,端粒会随着每次细胞分裂而缩短。当它们达到临界长度时,细胞就会进入衰老或凋亡状态。端粒酶缺乏与早衰疾病(先天性角化不良,dyskeratosis congenita)有关,而激活端粒酶则可以延缓小鼠的衰老[1:4]。
¶ 3. 表观遗传改变
衰老涉及表观遗传图谱的变化——即在不改变序列的情况下调节基因表达的 DNA 和组蛋白修饰。关键变化包括整体 DNA 低甲基化、CpG 岛的局灶性高甲基化,以及组蛋白修饰失衡(例如,H4K20me3 增加,H3K9me3 减少)。这些改变会导致转录噪声和异常的基因表达[1:5][3]。
¶ 4. 蛋白质稳态丧失
蛋白质稳态(protein homeostasis 或 proteostasis)确保蛋白质的正确折叠和清除。衰老会损害蛋白质稳态网络,导致错误折叠或聚集的蛋白质(例如 β-淀粉样蛋白、tau 蛋白)积累。关键机制包括分子伴侣失效和泛素-蛋白酶体系统功能障碍[1:6][4]。
¶ 5. 巨自噬失能 (2023 年新增)
巨自噬(Macroautophagy)是回收受损细胞器和细胞质内容物的主要机制。虽然最初被归类于蛋白质稳态中,但它现在被视为一个独立的标志。自噬随年龄增长而下降,导致“细胞垃圾”(脂褐素、受损线粒体)的积累。增强自噬(例如,通过雷帕霉素或亚精胺)在模式生物中能够持续延长寿命[1:7][5]。
¶ 拮抗性标志(对损伤的反应)
这些标志通常最初作为对原发性损伤的保护性反应,但当它们变成慢性或加剧时,就会变得有害。
¶ 6. 营养感应失调
营养感应通路网络——包括 mTOR、AMPK、Sirtuins 和 IGF-1——调节细胞的生长和代谢。在衰老过程中,合成代谢(生长)信号(mTOR/IGF-1)通常过度活跃,而分解代谢(修复)信号(AMPK/Sirtuins)则下降。热量限制和 mTOR 抑制剂(雷帕霉素)是已知最有效的长寿干预措施之一[1:8][6]。
¶ 7. 线粒体功能障碍
线粒体产生能量(ATP),但也会产生活性氧(ROS)。衰老会导致线粒体效率下降、生物发生减少以及 mtDNA 突变的积累。2023 年的更新强调,ROS 并非绝对有害,而是作为信号分子发挥作用(线粒体激效反应,mitohormesis);当这种信号传导被破坏或线粒体质量控制(线粒体自噬,mitophagy)失效时,就会出现功能障碍[1:9]。
¶ 8. 细胞衰老
衰老(Senescence)是由压力(例如,DNA 损伤、端粒缩短)诱导的稳定细胞周期停滞状态。虽然它对肿瘤抑制和伤口愈合至关重要,但衰老组织中衰老细胞的积累会通过衰老相关分泌表型 (Senescence-Associated Secretory Phenotype, SASP)——一种损害邻近组织的促炎细胞因子和蛋白酶的混合物——驱动病理变化[1:10][7]。
¶ 综合性标志(表型的罪魁祸首)
当积累的损伤和拮抗反应压垮组织稳态,导致功能衰退时,就会出现这些标志。
¶ 9. 干细胞耗竭
由于成体干细胞的消耗或功能障碍,组织的再生潜力随着年龄的增长而下降。这是由所有上游标志驱动的(例如,造血干细胞中的 DNA 损伤,卫星细胞中的衰老)。其结果是组织维持和修复失败,导致贫血、少肌症和免疫衰老[1:11]。
¶ 10. 细胞间通讯改变
衰老会破坏细胞间的通讯信号(内分泌、旁分泌和神经元)。这会导致一个“嘈杂”的系统环境,其特征是慢性炎症(炎性衰老)和神经-内分泌-免疫网络缺陷。像异体共生(年轻血液交换)这样的策略表明,恢复年轻的信号因子可以使衰老组织恢复活力[1:12]。
¶ 11. 慢性炎症 (2023 年新增)
通常被称为“炎性衰老 (inflammaging)”,这是一种随着年龄增长而积累的无菌、低度、持续的炎症。与急性炎症(一种防御反应)不同,慢性炎症会导致全身性组织损伤。它由衰老细胞的 SASP、细胞碎片 (DAMPs) 和免疫系统失调(免疫衰老)驱动。它是包括心血管疾病和神经退行性疾病在内的多重合并症的关键驱动因素[1:13][8]。
¶ 12. 菌群失调 (2023年新增)
肠道微生物组在衰老过程中会发生显著变化,失去多样性并向促炎特征转变(例如,Proteobacteria(变形菌门)的增加)。这种“菌群失调”(dysbiosis)会破坏肠道屏障(“肠漏”),使细菌产物进入血液循环并引发全身性炎症。恢复年轻的微生物组(例如,通过粪菌移植或益生菌)可以延长鳉鱼等模型动物的寿命[1:14][9]。
¶ 相互联系与治疗方法
这些标志并不是孤立的系统,而是一个复杂、相互作用的网络。例如:
- 基因组不稳定会引发细胞衰老。
- 衰老会产生 SASP,驱动慢性炎症。
- 炎症会损害干细胞功能。
- 自噬功能受损会阻碍受损线粒体的清除(线粒体自噬失败),导致进一步的炎症(炎症小体激活)。
这种相互联系意味着针对一个标志的干预通常会影响其他标志。例如,senolytics(清除衰老细胞)不仅能减少衰老,还能降低慢性炎症并改善干细胞功能[1:15]。
¶ 参考文献
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243-278. DOI: 10.1016/j.cell.2022.11.001 ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194-1217. DOI: 10.1016/j.cell.2013.05.039 ↩︎ ↩︎
Sen, P., Shah, P. P., Nativio, R., & Berger, S. L. (2016). Epigenetic mechanisms of longevity and aging. Cell, 166(4), 822-839. ↩︎
Kaushik, S., & Cuervo, A. M. (2015). Proteostasis and aging. Nature Medicine, 21(12), 1406-1415. ↩︎
Hansen, M., Rubinsztein, D. C., & Walker, D. W. (2018). Autophagy as a promoter of longevity: insights from model organisms. Nature Reviews Molecular Cell Biology, 19(9), 579-593. ↩︎
Fontana, L., & Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell, 161(1), 106-118. ↩︎
Gorgoulis, V., et al. (2019). Cellular senescence: defining a path forward. Cell, 179(4), 813-827. ↩︎
Franceschi, C., & Campisi, J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 69(Suppl_1), S4-S9. ↩︎
Ralli, C., et al. (2022). The gut microbiome and healthy aging. Nature Reviews Gastroenterology & Hepatology. ↩︎